LEYENDO GENES CADA VEZ MÁS RÁPIDO
¿Cómo se consigue secuenciar un genoma?
En poco más de una década hemos conseguido pasar de secuenciar a trancas y barrancas unos pocos genomas a producir de forma rutinaria una cantidad monstruosa de datos genómicos que los científicos apenas pueden asimilar, y el progreso sigue su avance gracias a la nanotecnología y a la disponibilidad de ordenadores cada vez más potentes.

Publicidad
Desde el descubrimiento de la estructura en doble hélice del ADN en 1953, los biólogos no han cesado en su empeño de conocer con detalle la estructura y funcionamiento del material genético, también conocido como genoma.
El genoma de cada ser vivo contiene básicamente las instrucciones del desarrollo y funcionamiento que el organismo en cuestión, desde una diminuta bacteria hasta el rorcual azul (el mayor animal de la tierra), necesitan para vivir. En principio cada una de las células contiene un genoma completo que emplea para la síntesis de moléculas y su regulación. La analogía con un libro, o con un texto en general, es muy conveniente, ya que en última instancia las moléculas de ADN son una sucesión lineal de elementos (bases o nucleótidos; miles de millones de ellos), y conocer este texto (secuenciar el genoma) es lo que nos abre las puertas al funcionamiento y evolución de los organismos.
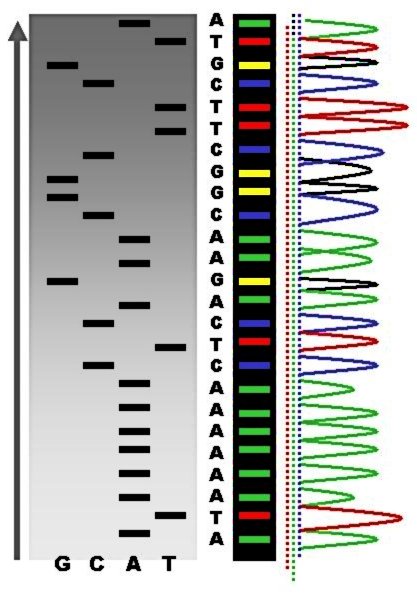
 ¿Cómo se secuencia un genoma? ¿Cómo se “lee” ese libro genético? A lo largo de las pasadas décadas se han ido sucediendo distintos sistemas, cada vez más sofisticados y eficaces. Originalmente la secuenciación se realizaba más o menos como leeríamos un libro: empezando en una parte del texto y continuando la sucesión de letras (o de nucleótidos) por orden. La capacidad de amplificar fragmentos concretos del genoma y secuenciarlos fue posible gracias al desarrollo de la PCR (Reacción en Cadena de la Polimerasa) y la secuenciación por electroforesis capilar, también conocida como Método Sanger. Dejando al margen los detalles, lo relevante de estas aproximaciones es que había que seleccionar una parte del genoma, secuenciarla, y sólo entonces se podía saltar a la región adyacente, siguiendo un “orden de lectura”.
¿Cómo se secuencia un genoma? ¿Cómo se “lee” ese libro genético? A lo largo de las pasadas décadas se han ido sucediendo distintos sistemas, cada vez más sofisticados y eficaces. Originalmente la secuenciación se realizaba más o menos como leeríamos un libro: empezando en una parte del texto y continuando la sucesión de letras (o de nucleótidos) por orden. La capacidad de amplificar fragmentos concretos del genoma y secuenciarlos fue posible gracias al desarrollo de la PCR (Reacción en Cadena de la Polimerasa) y la secuenciación por electroforesis capilar, también conocida como Método Sanger. Dejando al margen los detalles, lo relevante de estas aproximaciones es que había que seleccionar una parte del genoma, secuenciarla, y sólo entonces se podía saltar a la región adyacente, siguiendo un “orden de lectura”.
 Los primeros pasos de secuenciación de genomas fueron, desde la perspectiva actual, muy lentos. El primer genoma completo en secuenciarse fue el del bacteriófago FX174 en los años '70, un virus cuyo genoma consistía en apenas 5.000 de esas “letras” moleculares o bases.
Los primeros pasos de secuenciación de genomas fueron, desde la perspectiva actual, muy lentos. El primer genoma completo en secuenciarse fue el del bacteriófago FX174 en los años '70, un virus cuyo genoma consistía en apenas 5.000 de esas “letras” moleculares o bases.
Llevó bastantes años aún poder empezar a secuenciar genomas de organismos sencillos, como bacterias o levaduras (todavía siguiendo este sistema ordenado de lectura). Cuando en los '90 se inició el Proyecto Genoma Humano se hizo evidente que esta aproximación ordenada era extremadamente lenta.
La irrupción de la empresa Celera en esta odisea científica de la era moderna trajo consigo una posibilidad que puede parecer poco intuitiva, pero que resultó ser muy eficaz a largo plazo (y que fue adoptada de forma rutinaria): en lugar de leer ordenadamente el genoma, éste se fragmenta en millones de secuencias cortas y se leen por separado. Sería como hacer trizas el libro que queremos leer y limitarnos a reconstruir las frases de forma desordenada para luego acometer la tarea titánica de ordenar todos esos fragmentos (algo que sólo es posible gracias a potentes ordenadores).
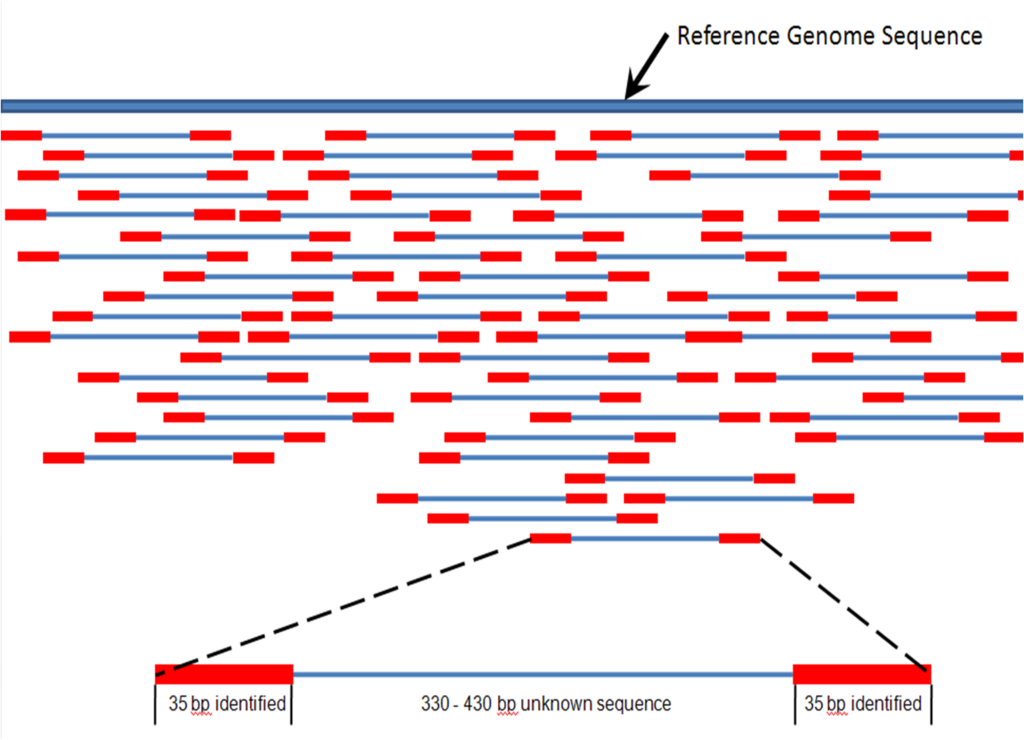
Fig 3. En la actualidad los genomas no se secuencian siguiendo un orden, sino que se fragmenta y se leen de forma desordenada y luego las secuencias se alinean por ordenador al detectar dónde se solapa cada fragmento. Fuente: Wikimedia.
De esta forma no sólo se consiguió acabar la lectura del genoma humano (de unas tres mil millones de “letras” genéticas), sino que estuvo acompañado, en los primeros años del siglo XXI, por un goteo de genomas relevantes para investigación por tratarse de organismos modelo, es decir, que son objeto de experimentación genética: los genomas de la mosca del vinagre (125 millones de bases), del ratón (tres mil millones de bases) o de Arabidopsis (una planta pariente de la col con 119 millones de bases). La investigación genética en estos organismos modelo es muy relevante en campos tan diversos como la medicina o la mejora agrícola de los cultivos.
Sin embargo, las técnicas de secuenciación aún no habían experimentado el gran salto que traerían los años siguientes.
A efectos de secuenciación de genomas completos, el método Sanger fue sustituido por las llamadas “técnicas de nueva generación” ('Next Generation Sequencing'), como la pirosecuenciación 454 o la plataforma Solexa/Illumina. Estos sistemas supieron aprovechar las ventajas de la nanotecnología para amplificar y secuenciar fragmentos de ADN en espacios mínimos, permitiendo la lectura simultánea de miles de millones de pequeños fragmentos de ADN al mismo tiempo. Baste un ejemplo:
Se tardaron más de diez años en secuenciar las tres gigabases del genoma humano. Uno de estos aparatos es capaz de secuenciar diez veces más ADN en un par de días. El desafío, hoy en día, es disponer de ordenadores suficientemente potentes para sintetizar y entender esos datos a un ritmo adecuado.
La gran ventaja de las técnicas de nueva generación es que han abierto la puerta al estudio genómico de organismos que no son modelos de investigación. Los costes se han reducido tanto en los últimos años (y parece que lo seguirán haciendo) que los biólogos evolutivos han empezado a secuenciar y comprar genomas de las más diversas procedencias: insectos, animales marinos, plantas y animales sin un interés económico inmediato (pero con una posición filogenética relevante).
El objetivo puede ser tan diverso como la reconstrucción de fenómenos evolutivos, la caracterización del conjunto de microorganismos de una muestra de una sola vez o la comparación entre poblaciones de una misma especie. No queda aquí la cosa: la progresión de nuestras capacidades secuenciadoras sigue en aumento, y ya se habla de una inminente tercera generación de secuenciadores que quién sabe hasta dónde será capaz de llevarnos.
Crédito de las imágenes pequeñas:
> Fig 1. Watson y Crick posando junto a un modelo de la doble hélice (Fuente: nature.com)
> Fig 2: Esta es la pinta que tiene una secuencia de ADN obtenida por el método Sanger (Fuente: Wikimedia)
Publicidad